Office of Research & Development |
 |
Talk to the Veterans Crisis Line now
An official website of the United States government
| Go back |
120 records found for:
VAEDA FAQ - VAIRRS System InquiriesDate Published: 2024-08-07 00:00:00.0 Recertification Date: 2025-02-07 00:00:00.0 Owner: VAIRRS
Question: Is there a recommended review or determination tool for the approving official to use in VAIRRS?
Answer: The VAEDA questionnaire and logic will eventually be translated into a wizard, iVAEDA, to support research determinations within VAIRRS. VAEDA will continue to function outside of VAIRRS for project leads not affiliated with VHA research.
| ||
ORD VAIRRS FAQ - RecordsDate Published: 2024-06-12 00:00:00.0 Recertification Date: 2024-08-11 00:00:00.0 Owner: ORD VAIRRS
Are local sites using IRBNet required to maintain hard copies of research records? For FDA-regulated studies? No, VAIRRS, VA's enterprise instance of IRBNet, has an Authority to Operate (ATO) to store VA records and is fully compliant with the FDA requirements at 21 CFR Part 11. The research administration office does not need to download the research records created and stored in IRBNet (VAIRRS) into another electronic storage location or hard copy subject to local and Sponsor policy. Are documents in IRBNet considered an official “record”? Yes. While IRBNet is a platform, it has an Authority to Operate (ATO) to store official VA records. There is no need for the research administration office to download records created and stored in IRBNet (VAIRRS) into another electronic storage location subject to local and Sponsor policy. | ||
ORD VAIRRS FAQ - ResourcesDate Published: 2024-06-12 00:00:00.0 Recertification Date: 2024-08-11 00:00:00.0 Owner: ORD VAIRRS
Is there an instruction manual that can be distributed for researchers and/or administrators? IRBNet is an intuitive, web-based tool that has instructions and help text available throughout the application. For additional assistance, users can reference various support resources within the VAIRRS University training library (VA network access only). The library is organized to assist with content navigation to help users access resources specific to their individual needs. Where can newly transitioned sites go for more help once training is complete? Many resources are available for newly transitioned sites. VAIRRS University (VA network access only) is an excellent resource that allows VA research employees to easily locate and access VAIRRS-specific training materials. You may also request a one-on-one mentor by emailing the project team at VAIRRS@VA.gov . Please make sure all committee administrators are included on the VAIRRS Administrators listserv to ensure receipt of monthly training announcements. | ||
ORD VAIRRS FAQ - Conflict of Interest (COI)Date Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Where in IRBNet can sites manage financial conflict of interest (COI) reviews? Sites can manage financial COI reviews on the VAIRRS SharePoint portal (VA network access only) and document reviews using the sample letter template, COI Acknowledgment Sample. | ||
ORD VAIRRS FAQ - CITI TMSDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Does IRBNet link to TMS and CITI? Yes, IRBNet supports the integration of TMS and CITI, so that records can be automatically created for users who link their IRBNet accounts to TMS or CITI. CITI integration is complete. TMS integration is in progress and is expected to be completed by early 2023. | ||
ORD VAIRRS FAQ - ePROMISEDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Will IRBNet update the station's ePROMISE records? Currently, the system does not integrate with ePROMISE. The plan is to integrate VAIRRS with other ORD enterprise systems in the near future. | ||
ORD VAIRRS FAQ - FA-SRDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Can you tell us more about the impetus for the Feasibility Alignment and Scientific Review (FA+SR) checklist? Users requested that the Feasibility and Alignment assessment be separate from the Scientific assessment, since many projects come with a scientific review. The form was created because so many facilities were not assessing feasibility until the end of the study, if at all. After thousands of dollars of review time conducted by committees, R&D committees were then challenging if the study could be done or altering the study, causing amendments to be filed with other committees, using even more committee time. Ultimately, the perceived non-completion rate, especially investigator-initiated studies, was the impetus. The burden on the research staff and committees to review projects that are not feasible or scientifically invalid is significant and takes time away from viable and scientifically valid studies. If the project does not meet the definition of research, does the FA or SR evaluation stop? Unless a researcher's facility requires their review, the research service is not responsible for projects that do not meet the research definition. The quality office is responsible for projects that involve quality improvement/assurance or evidence-based practice, etc. Can the FA+SR reviewer checklists be completed after submission to sub-committees, or is the expectation that R&D provides this review first and then repeats the evaluation after approval from the other committee? The review needs to be completed by someone (not necessarily an R&D member, though they likely have the best grasp of the projects happening across the facility) who can assess whether the project can be accomplished in the proposed time frame by the provider submitting it and if it will interfere with other studies happening at the facility. A statement from the investigator's supervisor stating they have the time and are qualified to conduct the project would go a long way in helping the person reviewing the form. The review is intended to be complete before it is assigned to any committee for review to reduce the burden on the committees of studies that are not feasible or scientifically valid. Is it acceptable if a site considers feasibility and scientific merit, but lacks a formalized Feasibility Alignment and Scientific Review (FA+SR) process? The use of the FA+SR checklists are encouraged, but not required if the facility has an established process in place. This process was established to help sites that do not have an existing FA+SR process. FA+SR is designed to determine whether the project has the appropriate resources and expertise to complete the study, whether the nature of the research is appropriate to be conducted at a given institution, and whether the project is sufficiently meritorious to proceed. The goal of FA+SR is to determine these factors at the beginning of the regulatory review process. Is the burden of FA+SR shifted from the IRB to the R&DC? The workload of the R&DC has increased substantially and seems unsustainable. FA+SR is an institutional responsibility and does not necessarily fall within the IRB's purview. The goal of FA+SR is not to increase the workload of the R&DC. The VA Electronic Determination Aid (VAEDA) is now available to all VA facilities. VAEDA is intended to relieve the R&DC and IRB from reviewing projects that are not research, thereby reducing the burden on both committees. The initial FA review can be conducted by an R&DC member and is intended to keep projects that are not feasible from ever reaching the R&DC. Like the Enterprise Research Data Security Plan (ERDSP), if proposed technology has little to no chance of being acquired or approved within the VA environment, then hopefully, the R&DC will not have to review the project. The goal of FA+SR is to determine these factors at the beginning of the regulatory review process. | ||
ORD VAIRRS FAQ - Forms TemplatesDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Will smart forms, fillable forms, wizards and letter templates be made available to the sites transitioning to VA's instance of IRBNet (VAIRRS)? ORPP&E, along with a field-based workgroup, has developed a core library of form and letter templates for each of the committee and subcommittee workspaces in VAIRRS. Users may preview the ORPP&E Standard Form Library (VA network access only) located within the VAIRRS SharePoint portal. The Project Cover Sheet wizard is required for all submissions. The IRB Information Sheet wizard is required for all submissions to the IRB (internal and external). A continuing review form, closure report and IACUC information sheet have been developed and will be available to all users. We are also converting the existing Microsoft Word forms to a fillable PDF format. Are sites required to use the ORPP&E core library? The ORPP&E core library was designed to provide a standard set of forms and letters for all sites. The Project Cover Sheet is required for all submissions. The IRB Information Sheet is required for all submissions to the IRB (internal and external). However, IRBNet supports the ability for you to upload your supplemental forms (such as if a particular form or template is required to support state or local regulations) via the Library Manager. Will each institution have specific templates for its forms? ORPP&E, along with a field-based workgroup, has developed a core library of form and letter templates for each of the committee and subcommittee workspaces in IRBNet. However, IRBNet supports the ability for you to upload your supplemental forms (such as a particular form or template required to support state or local regulations) via the Library Manager. If a change to an existing form is needed, fill out a change request form located on the VAIRRS SharePoint (VA network access only). For modified forms and newly created forms, the draft forms will be posted to the VAIRRS SharePoint portal in the ORPP&E Standard Form Library (VA network access only) for a review period. Notification for all new forms, wizards and letter templates will be included in the monthly VAIRRS newsletter. Does ORPP&E have guidance on how each smart form/wizard question should be answered? Researchers must answer simple “Yes” or “No” questions. ORPP&E reviews all feedback on the smart forms from the research community. If a question seems to be producing inconsistent answers or is causing confusion among your researchers, please email the project team at VAIRRS@VA.gov . Users can reference the VAIRRS IRBNet Wizards Navigational Guide (VA network access only) for high-level instructions on how to navigate the online wizards in IRBNet. Should the OGE Form 450 Alternative-VA be submitted through IRBNet? No, the OGE Form 450 Alternative-VA should not be uploaded into IRBNet. ORPP&E has requested that the researcher submit the OGE Form 450 Alternative-VA outside of IRBNet. The researcher may attach the findings to their package if the COI review is complete. The new financial COI IRBNet module was deployed in Summer 2022. Where do investigators, administrators and committee members obtain documents/templates in the ORPP&E Library outside of IRBNet? Investigators, administrators and committee members should refer to the ORPP&E Standard Form Library (VA network access only) on the VAIRRS SharePoint (VA network access only). The library is broken into three categories: Researcher/Administrator, Committee Members/Reviewers and Summary of CCB Changes. Once a category is selected, documents are further broken down to three folders: Forms & Templates, Letters, and Additional Documents. By selecting the correct library, users assure they are using the most recent template and alleviate the need to maintain a copy of the standard ORPP&E documents. Where do investigators obtain documents/templates in the ORPP&E Library? Investigators may access the ORPP&E library using the two methods described below. The research program does not need to maintain the standard ORPP&E documents in the local library. Method #1 Select “Forms and Templates” from the Other Tools menu. Select the appropriate VHA ORPP&E library. 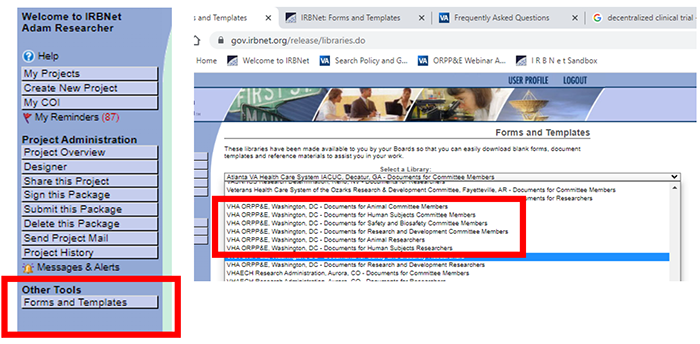 Method #2 On the Designer page, select the appropriate library in the “Select a Library” drop-down. 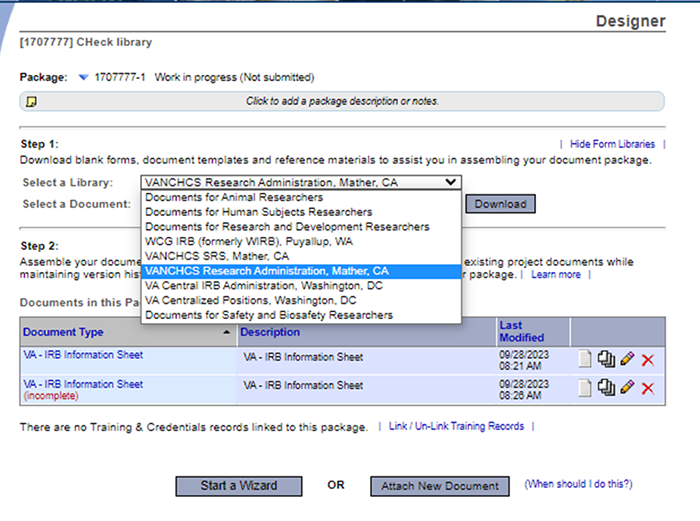
| ||
ORD VAIRRS FAQ - SignaturesDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Is the IRB Chair required to sign approval letters at initial or continuing review? ORD policy does not require that research approval letters be signed. However, there should be some means of ensuring the legitimacy of the letter and the approval date. In cases where an IRB or Privacy Board has approved a waiver of HIPAA authorization, the form must be signed by the IRB or Privacy Board Chair or a designated member of the IRB or Privacy Board. Is a wet signature required on board approval letters? No, there is no ORD policy requirement for a wet signature on any document. VHA policy allows for an electronic or digital signature wherever a signature is required. Local policies may restrict you to wet signatures where wet signatures are described in your local policy. Changes to local policies are recommended to accommodate electronic signatures. Can the certification statements within the Animal Component of Research Protocol (ACORP) be acknowledged with an electronic signature? Yes, the certification statements in the ACORP may be signed digitally (defined by VA Handbook 6510 as a specific electronic signature technology that allows the recipient to prove the origin of the document and to protect against forgery) or with a physical (wet ink) signature - whichever is preferred. What is important to ORD is that the signature is personally applied by the signatory, unique to the signatory, dated and unambiguously associated with the certification statement on the final approved version of the ACORP. A signature on the IRBNet package does not apply to any particular certification statement, and signatures provided only at the time of initial submission do not document acknowledgment of the contents of the final approved version if the IACUC required any changes before approval. To meet VA requirements, the investigator may digitally sign the ACORP using Adobe Reader and upload the signed ACORP to the package in IRBNet for review. If the IACUC requires modifications before approval, the investigator must also sign the revised version. After the ACORP has been approved with no further modifications, the certification statements on the final approved ACORP (already signed by the investigator) must be signed by the IACUC chair, attending veterinarian and any others whose signatures are required for applicable appendices. The IACUC administrator should publish the approved, signed ACORP as a board document. Please note that VA now requires the signatures in the ACORP for every protocol documented on the ACORP form (see Guidance Document AR2020-001: https://www.research.va.gov/programs/animal_research/guidance.cfm ). Are coordinators required to sign every package they submit? Yes, every package must be signed in IRBNet using the “Sign this Package” feature by the principal investigator (PI) or by the submitter on behalf of the PI. | ||
ORD VAIRRS FAQ - Skype for BusinessDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Our site has installed the “Skype for Business” add-on for Internet Explorer. Can we use this add-on with IRBNet? There is a known issue with the “Skype for Business” add-on for Internet Explorer that affects users who create rich-text content that includes phone numbers. In IRBNet, this primarily affects administrative users who draft decision letters in the rich-text editor. Microsoft has not yet released an update to address this issue. If possible, we recommend that staff who edit letters disable the add-on or use an alternate browser. | ||
ORD VAIRRS FAQ - SubmissionDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Is there a flow chart of the VAIRRS submission process that can be shared with the IRB, RDC, Safety, IBC,IACUC committee members and researchers? Yes, a high-level flowchart of the intended submission process is available for download within the VAIRRS University training library (VA network access only). | ||
ORD VAIRRS FAQ - Animal ResearchDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Are wizard forms applicable to animal studies? The Project Cover Sheet wizard is required at initial submission for all studies (animal studies included).The IRB Information Sheet wizard is not required for animal studies. Does the IRB Information Sheet need to be filled out for animal research? No, the IRB Information Sheet wizard is only required for studies submitted to the IRB. How should VA IACUCs record their facility number and address into the IRBNet Minutes Builder to meet the 1200.07 directive requirements? VA IACUCs should enter their facility number and address into the Agenda Location field in IRBNet. ORD and ORO have determined that if the facility number and address are entered into IRBNet in this manner that will satisfy the requirement from the 1200.07 directive | ||
ORD VAIRRS FAQ - Maintenance of a PackageDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
If a package has been unlocked, how does a committee maintain the integrity of a package? If a package is incomplete and requires additional documents to be ready for review, it may be unlocked and returned to the study team prior to the committee's review. Once the package has been shared with a reviewer, it should not be unlocked. If the committee determines that modifications are required to secure approval, then the modifications or response materials should be submitted in a new package to preserve the original documents. Do older studies need to complete the Project Cover Sheet wizard? Older active studies uploaded when the site enrolled in IRBNet should complete the Project Cover Sheet wizard as soon as possible. The IRB Information Sheet wizards should be completed at the next continuing review (CR). When completing a wizard, what should be done if the package has already been sent along for PO, ISSO and committee review? Previously reviewed projects should have a completed Project Cover Sheet or IRB Information Sheet. However, when submitting wizards - specifically in response to the ORD Data Call - approved studies may disregard the guidance text published at the end of the wizard. Is there a process for handling duplicate projects for studies overseen by the CIRB? Can duplicate projects be merged or deleted? Projects with a CIRB shell and a duplicate local project may be reconciled if there has been no submission using the CIRB shell. Please get in touch with VAIRRS@va.gov for further instructions. Does the VAIRRS system allow investigators to submit a package directly to a committee? Currently, the system does not allow a package to be submitted directly to a committee. An administrator should review a package for completion before sending it to a committee. Can a package be unlocked for a single committee, but locked for other committees? Do packages operate independently? No, if a committee unlocks a package, it is unlocked for all committees. Once a package has been submitted, is it possible to update package forms and documents? Packages are automatically locked upon submission. The researcher would only be able to modify documents in a package after submission if a committee unlocks the package. They can, however, always submit revised or updated documents in a new package. How does a user update a study in IRBNet if staff leave the project? For studies under the oversight of the Central IRB, users can list staff that have left the study in 115 section II. Section II has various areas in the 115a PISC Continuing Review Application and the 115b Local Site Investigator (LSI) Continuing Review Application to list staff that have left the study. For non-Central IRB studies, please follow local guidance for required form updates. All studies must update the personnel section of the Project Cover Sheet wizard to remain consistent with current staffing. How can I update a principal investigator (PI) for a project in IRBNet? The PI or a study team member with full access can change the PI in the next package by selecting the “Edit” button on the Project Overview page. The PI's name cannot be changed on previously submitted packages.Users can reference the PI Change Instructions User Guide for further guidance (VA network access only). In IRBNet, when a study is renewed, should the initial approval date be changed to the most recent renewal date? The initial approval date represents the date the project received its first approval from the oversight board. The initial approval date should not be changed or updated when recording renewals. | ||
ORD VAIRRS FAQ - VAIRRS System InquiriesDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Do committees have the same set of keys as IRB administrators? The committee and administrator tools may differ depending on the user's role and level of access. What is VAEDA (VA Electronic Determination Aid)? VAEDA is a new, innovative system, available to all VAMCs, created by the ORD to standardize and streamline processes within VA research. VAEDA is a decision support tool created to reduce variability in regulatory determinations for proposed research, quality improvement, program evaluations and innovation projects. To learn more about VAEDA, click here . Is VAEDA an IRBNet update? No, VAEDA is independent of IRBNet. Am I allowed to use a non-VA email address as my recovery email in IRBNet? Yes, ORD recommends that all IRBNet users utilize a non-VA email address as their recovery email to help ensure continued seamless access to IRBNet when not logged in to the VA network. Your recovery email is used to receive the multi-factor authentication (MFA) code sent to you when you access IRBNet for the first time from a new device. Since IRBNet generates this code, this code is not VA data, and receiving this code to your non-VA email address does not violate VA policy. How can I view a researcher's current training? There are several ways to get this information. The first option is to use the Track Training tool. When searching for a user in the Track Training tool, select Set Dates as of Today. This will pre-populate your search criteria so that it only looks for current trainings. The second option is to use the Project Team Tracking section of the Submission Detail page. This section lists all of the researchers on the project and any accepted training records for types that you have flagged. For example, if a researcher is required to take a given CITI course, then you can flag that course on the Manage Types and Flags page, and any time you open the Project Team Tracking section, any researchers that have current training for that course will have a colored flag next to their name. The third option is to use a Training Insight Report. This will give you all of the training records accepted by your research program, which you can then filter for current training. This last option is primarily helpful for reporting across the entire program, instead of an individual project (such as, if you want to know how many people have taken a given course). Where do we get the VA Central IRB study number (the 4-digit number)? The number (two-digit year-sequency number) is assigned by the VA CIRB office and is added to IRBNet once it is assigned. The number is inserted as the Ref # next to the submission date on your Submission Manager page. How does a new staff member gain access to IRBNet? New staff members must register through lRBNet . The local IRBNet administrator must send an email to IRBNet Support at govsupport@irbnet.org for new staff members to request administrator access to the appropriate workspace(s). For user instructions on how to register, users can reference various support resources within the VAIRRS University training library (VA network access only). Are research coordinators able to see documents associated with a project? If a project is shared with a user, the user should have read access to all documents associated with that project. Read access allows the user to view the documents associated with a project. However, the user will not be able to modify documents or submit a package. Is the Submission Manager only available to IRB administrators? The Submission Manager is available to administrators and reviewers. It is not available to study coordinators or study PIs. If a facility does not have a Submission Manager and thinks they should, please reach out to the IRBNet Support contact at your facility or email govsupport@irbnet.org . For a Central IRB study, how does a PI submit a package or project only for local review (not for the lead site or other sites to review)? A PI or study team member should create a new package within the project and submit directly to the research committee at their particular site, instead of to VA Central IRB administration. When setting up studies in IRBNet, should all study team members be listed? Yes, all study team members should be shared on the project to ensure their training records are linked. How can a user request IRB reports to be generated and delivered monthly? Administrators can email govsupport@irbnet.org to request IRBNet insight reports. How does one access the Field Staff Dashboard? VAIRRS has published Power BI Dashboards for every medical center in the VA system. To request access to the Field Staff Dashboard, fill out the request form (VA network access only) located on the VAIRRS SharePoint site. How are new user requests processed through IRBNet? If the individual is a new user, they can go to VA's instance of IRBNet, VAIRRS, and create an IRBNet account. If the individual is working to gain access for a research office staff member at their site, then the local IRBNet administrator would email IRBNet Support ( govsupport@irbnet.org ) to request the individual be granted access to the relevant workspace(s). | ||
ORD VAIRRS FAQ - Project Cover SheetDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
For multi-site studies, to which site should the Project Cover Sheet be submitted? For studies under the oversight of the VA Central IRB, the Project Cover Sheet should be submitted to the VA Central IRB for the lead site and each local site. For multi-site studies with a Central IRB shell and a duplicate local project, is the Project Cover Sheet required for both the Central IRB and local versions? No, we are reconciling the list of active projects to account for studies with two versions in IRBNet.The Project Cover Sheet should be submitted to the Central IRB using the Central IRB shell. How do I close a project in IRBNet?
What is the purpose of the Project Cover Sheet (PCS) wizard? The PCS wizard is what drives a majority of the metrics needed for the research enterprise dashboards. The data collected in the wizard feeds the local and ORD dashboards.The Chief Research and Development Officer (CRADO) currently utilizes the ORD-level dashboard to view research activity at a national level. The facilities participating in the Dashboard Focus Group may access their dashboard to view research activity at their facility. Dashboards for all medical centers were deployed in May 2022. The ORD dashboard is a decision-making tool that allows ORD to identify gaps in the national research program. Prior to VAIRRS, ORD did not have a central data collection tool that included data on ORD-sponsored, externally funded and locally funded projects. VAIRRS now provides a complete picture of research activity across the VHA research enterprise. Numerous groups within the Central Office are partnering with ORD to use the data provided by VAIRRS to decrease the burden on research offices as it relates to reporting. The end goal is to no longer need to conduct data calls out to the field. If a researcher submits a Project Cover Sheet wizard and information is found to be incorrect, can the research team go back to the wizard and correct the information? How can this be accomplished without having to generate a new wizard? The researcher has two options. First, if the Project Cover Sheet wizard is already submitted and the package is locked, the researcher can clone a previously completed wizard and jump to the section that needs to be revised. The second option is if the package is unlocked, and the researcher was instructed to revise it. There is a pencil icon next to the wizard on the “Designer” page for the package to allow them to edit the wizard for resubmission. Which study team personnel should be included in the Additional Personnel section of the Project Cover Sheet wizard? At a minimum, the principal investigator, study coordinator, and study team members for whom training is tracked in IRBNet or require access to the electronic health record (EHR) should be included in the study team section of the Project Cover Sheet. The personnel information provided in the Project Cover Sheet wizard should be consistent with any local personnel form that is required for the study action. On the “Project Personnel” page within the Project Cover Sheet (PCS) wizard, how do users utilize the “Adding, Removing, Editing or No Change” functionalities? The Adding, Removing or Editing question in the PCS enables users to specify if new personnel have been added to a study, if personnel have been removed from a study and/or if other personnel information needs to be updated. If none of these options are applicable (i.e., no changes are needed in this iteration of the form), users may select No Change. Users completing the PCS wizard for the first time should select the “Adding” option to list personnel. The data collected in this section will also be used by the ORD Strategic Initiative for Research & EHR Synergy (OSIRES) team to provision access at the time a user's VA medical center (VAMC) transitions to the Cerner Electronic Health Record. Please note that so long as an individual is listed on the PCS as “additional personnel,” their name and information will appear on the PDF, regardless of the option selected for the Adding, Removing or Editing question. An individual will only disappear from the PDF once they have been entirely removed from the form via the red X icon located at the top left of their name on the Additional Project Personnel page. Alternatively, while editing the PCS, a user may indicate to the board that they are adding a new individual to the study by selecting the Adding option. The new individual and their information will appear on the PDF. If a user has previously entered user information (i.e., a new project role) that needs to be updated on the PCS, the user may select the Editing option to update that individual's information. Whenshould the Project Cover Sheet – Additional Project Personnel section be updated if the change does not require an amendment? The Project Cover Sheet (PCS) Additional Project Personnel section must be consistent with local requirements and must be kept up to date with project amendments. As electronic health record (EHR) access is required to be changed, the PCS Additional Project Personnel section must be updated to ensure continuation of appropriate EHR access. If a study team member change does not require an amendment, the Additional Project Personnel section should still be updated to reflect the study team member change and submitted to their local research administration in IRBNet. PCS updates for study team member changes that do not require an amendment must not include changes to study details (i.e., study characteristics), as additional changes may require an amendment and oversight committee/subcommittee review. A user must follow the guidance of the oversight committee and/or subcommittee(s) for reporting changes in personnel that do not require an amendment. Please note, all study team members should be included in the “PCS Additional Project Personnel” section and shared on the IRBNet project to ensure training can be tracked in IRBNet. | ||
ORD VAIRRS FAQ - Study ReviewDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Within IRBNet, some CIRB studies have an expiration date, but no report due date; some have a report date, but no expiration date; and some have neither of those dates. What does this mean for each project? An expiration date indicates that continuing review is required. Reminder notices will go out at 90, 60 and 30 days prior to the expiration date. The continuing review is due into IRBNet 60 days before the expiration date. “Report due date” only means that an Annual Status Report is due from the PISC Study Team. The Annual Status Report is due 30 days before the due date. No dates would indicate that a local site does not require continuing review. How do we know which studies will require continuing review versus just the annual report? The initial approval letter will state whether or not continuing review is required. Your initial approval letter will note that continuing review is unnecessary if the study is minimal risk, adheres to the 2018 Common Rule and meets all other criteria. For studies under the oversight of the Central IRB, the approval letter will be specific and provide the individual with the form number: Form 130 Annual Status Report. Some studies are minimal risk and typically do not require continuing review; however, should the Central IRB reviewer or the Central IRB want to see that study, it will be stated in the initial approval letter and continuing review approval letter if an additional continuing review is needed. This is true even if only for one cycle of continuing review. | ||
ORD VAIRRS FAQ - RCO WorkspaceDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
Why did ORO decide to suddenly develop the VAIRRS/IRBNet RCO Workspace? ORD and ORO started discussions in July 2021. The VAIRRS contract modification was awarded in April 2022 and formally announced shortly thereafter. The VA contracted for the RCO module in response to feedback from RCOs that publishing audit results, notifying committees and researchers of audit results, and aggregating the data for the Facility Director Certification report was a burden on RCOs. Is the use of the VAIRRS/IRBNet RCO Workspace required? For IRBNet to provide an RCO with the aggregate data to support the FDC Part B, the RCO must record all of their audits for the reporting cycle within IRBNet using the electronic tools. Any mandate requiring the use of IRBNet would come from ORO. Will the new module create more work for the RCOs? No, the RCO module is expected to reduce the amount of work required by RCOs to record and share audit results in IRBNet. What are the benefits of the RCO module? RCOs will be able to easily and automatically access the electronic records they need, record audit results using the electronic audit tools, and automatically have VA committees and researchers properly notified and provided with access to the audit results. At the end of the 2022-23 reporting cycle, IRBNet will provide RCOs with aggregate data supporting that cycle's Facility Director Certification (Part B) report. Will RCOs be required to use the new module? For IRBNet to provide an RCO with the aggregate data, and for the data to be accurate, the RCO must record all of their audits for the reporting cycle within IRBNet using the electronic tools. The oversight committee may request RCO audits be submitted using RCO module. Use of the new moduleis strongly encouragedby ORO. Are projects required to be shared with all project team members? As part of each review process, the subcommittee must ensure that the project has been shared with all project team members. This is important because it affects an RCO's ability to access the complete project record and training information when performing an audit. Who has access to the RCO audits within VAIRRS? RCOs have the capability to publish an audit report internally or externally. When published internally, the audit report is only available to the individual that published it and other auditors from that site. When published externally, the audit report is available to the study teams and review committees. Publishing externally triggers an email notification to the study team and review committee. Sites can reference the IRBNet Notes – Enterprise Audit Tools (VA network access only) to address any other questions surrounding publishing. | ||
ORD VAIRRS FAQ - VAIRRS UniversityDate Published: 2024-05-23 00:00:00.0 Recertification Date: 2024-07-22 00:00:00.0 Owner: ORD VAIRRS
What is VAIRRS University? VAIRRS University is a revised training library designed to promote self-education and independent learning. This new platform allows VA research employees to easily locate and access VAIRRS-specific training resources. The library contains a variety of materials, best practices and tutorials developed by the VAIRRS Support Team and other VA medical centers (VAMC) that work within VAIRRS, as well as content generated by IRBNet and courses from VA's Talent Management System (TMS). How can VA research employees access VAIRRS University? VA research employees can access the VAIRRS University training library on the VAIRRS SharePoint portal by selecting VAIRRS University from the menu on the left-hand side of the page. How can users access training resources relevant to their role within VAIRRS? VAIRRS University is organized to assist with content navigation and to help users access resources specific to their individual needs. Users can select from seven user role pathways and five training material categories to view curated selections of resources available to them.
Who manages VAIRRS University? The VAIRRS Support Team manages VAIRRS University, updating existing content and adding new content, as needed, to enhance and improve the learning experience for VA research employees. Who can answer questions about VAIRRS University? Please contact the VAIRRS Support Team at VAIRRS@va.gov with any questions about VAIRRS University. | ||
Toolkit Research Laboratory Biosafety and BiosecurityDate Published: 2024-04-04 00:00:00.0 Recertification Date: 2024-10-04 00:00:00.0 Owner: Biosafety and Biosecurity
Frequently Asked Questions/FAQs
Guidance Documents
Templates
Webinars | ||
Toolkit RD CommitteeDate Published: 2024-04-04 00:00:00.0 Recertification Date: 2024-10-04 00:00:00.0 Owner: ePROS
Frequently Asked Questions/FAQsGuidance Documents
Memorandums
Templates - (Templates are located in IRB Net, or VAIRRS Standard Form Library)
Webinars
| ||
Toolkit Exempt ResearchDate Published: 2024-04-04 00:00:00.0 Recertification Date: 2024-10-04 00:00:00.0 Owner: ePROS
Flow Charts - (Flow charts can be found in IRB Net, or VAIRRS Standard Form Library)
Memorandums
Templates - (Templates can be found in IRB Net, or VAIRRS Standard Form Library)
Webinars
| ||
ePROS FAQ - Technology Transfer Program (TTP)Date Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Where can I find more information on the VA Technology Transfer Program (TTP)? I am interested in learning more about intellectual property. Answer: More information is found on their website at VA Technology Transfer Program. Questions can be directed to the following groups within TTP:
| ||
ePROS FAQ - Technology Transfer ProgramDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Where can I find Technology Transfer Program (TTP) templates and policy documents? Answer: TTP CRADA templates and some policy documents are available on the VA intranet only. If you have VA access go to their SharePoint site: VA Technology Transfer Program. | ||
ePROS FAQ - Proactive CallingDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Where can I find information on proactive calling? Answer: Calling potential VA subjects without first sending a notification in person or through mail or email for the purpose of VA research recruitment is NOT permitted outside of this ORD approval process unless there is written documentation that the subject is willing to be contacted by telephone about the study in question or a specific kind of research as outlined in the study as per VHA Office of Research and Development (ORD) policy in VHA Directive 1200.05, Requirements for the Protection of Human Subjects in Research. The acting Under Secretary of Health (USH) signed a policy memorandum on June 29, 2021 allowing for the proactive calling of VA subjects, sometimes referred to as “cold calling”, as a recruitment strategy for VA human subject research studies using an ORD approval process. Any request for proactive calling for subject recruitment in a VA study must be submitted and approved through this ORD process. ORD approvals are specific to the individual research study submitted with the request. More information about proactive calling and how to get ORD approval can be found here: Proactive Calling for Subject Recruitment - Home (sharepoint.com) | ||
ePROS FAQ - ORDs Biosafety Biosecurity ProgramDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is ORD's Biosafety & Biosecurity Program, where can I find more information, and who do I contact? Answer: The VA Office of Research and Development (ORD) Research Laboratory Biosafety & Biosecurity Program promotes the safe use of infectious agents, pathogens, toxins, recombinant and synthetic nucleic acids (collectively referred to as biohazards) in VA biomedical research programs. Biosafety is the consistent application of safety measures to minimize or prevent exposure to the person handling biohazards, lab and building occupants, the community and the environment. Biosecurity is the risk- and threat-based control measures established to prevent the unauthorized access, misuse, loss, theft, diversion and intentional release of valuable biological materials, pathogens, toxins, information, expertise, equipment, technology and intellectual property that have the potential to cause harm to humans, animals, plants, the environment, public safety or national security. ORD's biosafety and biosecurity program functions in coordination with similar programs at individual medical centers. ORD’s biosafety and biosecurity program’s activities involve multiple components, including developing VHA policy, issuing guidance, communicating training requirements and initiating training programs; it also involves coordinating with other Federal entities regarding biosafety, biosecurity and homeland defense. The program provides advice and guidance to VA Facility biomedical research programs on biosafety and biosecurity program management practices and strategies to control the risks associated with the respective biomedical research activities at field sites. More information can be found on the ORD website: Biosafety & Biosecurity Program (va.gov) Questions, comments, and suggestions regarding VA’s biomedical and biosafety research program or activities may be submitted to VHACOORDBiosafety@va.gov. | ||
ePROS FAQ - Invention Disclosure and CertificationDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How do I disclose an invention that I created as a VA employee? Answer: Invention Disclosure and Certification: Any invention made by a VA employee including, WOCs and IPAs, must be disclosed to VA, no matter how the invention came into being. VA inventors must each submit an Invention Certification statement for each invention they contribute to. The Invention Certification will be considered in determining whether the government is entitled to an interest in the invention. More information on how to disclose using TTP's Sophia online portal can be found here: Invention Disclosure and Certification (va.gov). | ||
ePROS FAQ - REDCapDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is VA REDCap? I'm interested in creating a database from surveys, will VA REDCap be an option to help? Answer: REDCap allows for easy creation of online databases and surveys without requiring knowledge of programming language. VA REDCap users can develop data collection instruments and control how the project is to be shared with others. When survey capability is enabled on an instrument, anyone with access to an internet connected device can respond to the survey. Other benefits include data quality control, secure storage, maintenance and sharing of data on the REDCap server, and consistent formatting across all sites with validation and branching/skipping logic. VA REDCap is approved for storage of PHI and PII, including patient name, social security number, date of birth, mother's maiden name, personal mailing address, personal phone number, personal fax number, personal email, emergency contact (name, phone), current medications, previous medical records, race/ethnicity, medical record number, dates of care, images (photograph, diagnostic image), geographic data, biometric identifiers, and any unique identifying number or code. Find more information, please visit the VIReC SharePoint site: VA REDCap. | ||
ePROS FAQ - DocuSignDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I am interested in using DocuSign to collect consent and HIPAA authorization from veterans in a rural community. How do I get started with that process? Answer: ORD has purchased a supply of envelopes to be used in research studies requiring documentation of informed consent and or HIPAA Authorization. Approval for use of DocuSign is limited to the VA’s contract with DocuSign. Only the VA’s contract with DocuSign managed by the Identity Access Management (IAM) Program Office has been approved with an Authority to Operate (ATO) to allow electronic signatures for documenting informed consent for VA research studies. | ||
ePROS FAQ - VA BoxDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I may be interested in using VA Box to share large imaging files with a university affiliate. What exactly is VA Box and how can I get an account set up? Answer: What is VA Box? VA Box enables easy access to content and collaboration tools uniting VA study team and non-VA colleagues with security, scalability, and administrative controls. From sharing and accessing files to sophisticated business processes like data governance and retention, VA Box is used to manage, share, and collaborate on sensitive and non-sensitive data files with people outside of the VA firewall. Acceptable Use Policy (AUP) How can I get APPROVAL & FUNDING? Office of Research and Development (ORD) will fund research related projects and welcome requests four (4) weeks before the need to begin sharing information with affiliate or other colleagues, and will consider the following types of requests:
What USES ARE NOT ALLOWED?
This does not mean that you cannot use VA Box.com. You may request that your department/sponsor fund the use of Box.com. More information can be found here. To submit a request or ask questions, use the following link: BOX|Digital VA Request a VA Box account from ORD at this SharePoint site: Home - VA Box.com Requests (sharepoint.com) | ||
ePROS FAQ - VA Box and QualtricsDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is it possible to use VA Box with Qualtrics? Answer: Yes! VA Box enables easy access to content and collaboration tools uniting VA study team and non-VA colleagues with security, scalability, and administrative controls. From sharing and accessing files to sophisticated business processes like data governance and retention, VA Box is used to manage, share, and collaborate on sensitive and non-sensitive data files with people outside of the VA firewall. Acceptable Use Policy (AUP) Qualtrics is an cloud-based platform for creating and distributing web-based surveys. To use them together see the training at this link. Email the VABoxTeam@va.gov for help. | ||
ePROS FAQ - Qualtrics Survey ToolsDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I am interested in conducting a Qualtrics survey, but am not sure how to get started. Do you have guidance? Answer: Qualtrics information can be found at this SharePoint site: VHA ORD Qualtrics Portal - Home (sharepoint.com)
| ||
ePROS FAQ - Support for professional and administrative servicesDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I need help funding research staff at my local VA research office. What are the parameters of duties as well as the scope and breadth of the support provided by ORD? How can I find more information and request help? Answer: Aptive Health Transformation Group (HTG) has been awarded a contract to provide support for VA Research Studies and Clinical Trials at VA Research Centers across the US and its territories. The purpose of this contract is to enable VA Research staff to request and receive support for professional and administrative services. Services to be performed include but are not limited to research study and clinical trial coordination, research administrative services, proposal development (specific to studies/trials), administrative services, research project management support, and research education and training support. Scope and breadth of requests may involve single-site, multi-site, or remote (off-site) research studies / clinical trials. Make a request for this service: Field Enterprise Research Support Services (FERSS) - Home (sharepoint.com) | ||
ePROS FAQ - Single IRB ExceptionsDate Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I am interesting in learning more about single IRB exceptions and would may want to make a request for an exception. Any advice? Answer: Answers to most questions about the process or policy of single IRB exceptions can be found at: Single IRB Implementation in VAs. To make a request for a single IRB exception, please visit this SharePoint site: ORD Single IRB Exception Requests - Home (sharepoint.com).
| ||
ePROS FAQ - Finding Information on the VA Central IRB (VA CIRB)Date Published: 2023-09-26 00:00:00.0 Recertification Date: 2024-08-16 00:00:00.0 Owner: ePROS
Question: How do I find more information on the VA Central IRB? Answer: More information can be found at their website: VA Central Institutional Review Board (IRB) Additionally, contact information can be found here: VA Central IRB Administration | ||
ePROS FAQ - IBC Meeting MinutesDate Published: 2023-09-07 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are the names of IBC protocol reviewers required to be included in the IBC meeting minutes? Answer: No. the names of IBC reviewers are not required to be included in the IBC meeting minutes by either VHA Directive 1200.08 (1) or the NIH Guidelines on Research Involving Recombinant and Synthetic Nucleic Acid Molecules. Additionally, ORD concurs with the NIH Office of Science Policy (OSP) FAQs about IBC Meetings and Minutes located at FAQs About IBC Meetings and Minutes - Office of Science Policy (nih.gov) that IBC minutes do not need to be transcripts or kept at a level of detail that attributes each remark to a specific individual. | ||
ePROS FAQ - Research Law Explorer- State and Tribal Law DatabaseDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Where can I find information regarding state and tribal laws applicable to VA Research? Answer: The Research Law Explorer (Explorer) helps inform VA investigators and study coordinators of state-level research laws. The Explorer allows users to locate, read, and export the most relevant and up-to-date state laws that may impact VA studies. The Explorer only provides publicly available legal information and in no way makes interpretive judgements about the laws presented. If users have questions about their research, they should still contact the Office of General Counsel. Users should not treat the Explorer as a primary reference for all state research laws. Users are responsible for conducting independent research and drawing their own conclusions with all the resources at their disposal. If a user believes that the Explorer is missing information, please report it using the blue feedback button at the top of this application. | ||
ePROS FAQ - Welcome to the VA Innovation and Research Review System (VAIRRS) SharePointDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is VAIRRS and where can I find information about it? Answer: The VA Innovation and Research Review System (VAIRRS) is the VA's enterprise instance of IRBNet. VAIRRS will be used by all VA medical centers with research programs and will provide an enterprise platform to support the management of research oversight committees. The transition to this new online submission and review system began in October 2020. Currently, VAIRRS supports 106 VA research sites. VAIRRS Mission: The VA Innovation and Research Review System (VAIRRS) supports management of the research enterprise and processes of research-related stakeholders in VA. VAIRRS Vision: VAIRRS seeks to be the trusted review platform and data source for all research-related stakeholders Find more information on the ORD SharePoint page. You must be logged into the VPN to access all ORD SharePoint pages. | ||
ePROS FAQ - HSRD Centers of Innovation (COINs) and Resource CentersDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What are the HSR&D Centers of Innovation (COINs), Resource Centers, and the Centralized Service Program and where can I find information about them?
Answer: There are 18 HSR&D Centers of Innovation (COINs), 4 Resource Centers, and 1 Centralized Service Program located throughout the US. VA's Health Services Research and Development Service (HSR&D) funds eighteen Centers of Innovation (COINs). The COINs will build on the successes of HSR&D's earlier Centers of Excellence (COEs) and Research Enhancement Award (REAP) programs. The COIN program rewards research innovations and partnerships to ensure that research has the greatest possible impact on VHA policies, healthcare practices, and health outcomes for Veterans. A unique feature of the COINs is that they include one or more focused areas of research that addresses questions of significance to VHA clinical and operational partners, and these partners will be engaged in the research activities of the COINs. Resource Centers and Centralized Service Program provide support to central office leadership and field investigators by disseminating HSR&D research findings; providing information and consultation about VA data; and connecting researchers with each other through ongoing education and symposia. More information can be found on the HSR&D website. | ||
ePROS FAQ - VA ORD Research ProgramsDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Where can I find information on ORD Research Programs, Centers, and REAPS? Answer: Details on each of the Programs, Centers, and REAPS can be found below: | ||
ePROS FAQ - Local Research Office Contact VerificationDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: I am a site liaison for the VA Central IRB. We have filled a position in our local research office. How do we let the CIRB/ORD know of this update? Answer: Please go to the Verification Instructions on ORD's SharePoint page. Please note that only Site Liaisons are able to update this page. If you are a new site liaison and cannot access this list, please contact the VA Central IRB at vacentralirb@va.gov. | ||
ePROS FAQ - VA Electronic Determination Aid (VAEDA)Date Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is VAEDA and where can I find more information on it? Answer: The VA Electronic Determination Aid (VAEDA) is a new, innovative system created by the Department of Veterans Affairs (VA) Office of Research and Development (ORD) to standardize and streamline processes within VA research. VAEDA is a decision support tool created to reduce variability in regulatory determinations for proposed research, quality improvement, program evaluations and innovation projects. Among the many benefits of this new system, VAEDA is guaranteed to save research staff valuable resources, including time, money and energy. By consolidating information in a centralized platform and providing research determinations through a systematic, scientific approach, VAEDA eases the burden of research processes in VA facilities across the country. Mission: VAEDA serves as the enterprise solution for research determination management and classification within VA.Vision: VAEDA provides researchers with services for expedited, standardized determinations and will be appropriately positioned to evolve into “iVAEDA” as two innovative technologies become one interconnected software built to benefit the VA research community nationwide.
| ||
ePROS FAQ - DocuSign RetentionDate Published: 2023-08-31 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is the DocuSign document retention policy? Answer: Six months. DocuSign Cloud is not a place to store study documents indefinitely. Once a document is completed or signed, it must be downloaded to wherever all other study files are stored. If a document that has been signed is older than six months old, it will be deleted. It is the investigator’s responsibility to make sure all study documents are stored someplace other than the DocuSign Cloud. | ||
ePROS FAQ - VHA Research Activities withing the Paperwork Reduction ActDate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What types of research activities conducted by VHA are not required to apply the PRA according to the Cleland-Dole Act? Answer: ORD’s policies define research for any research activity conducted within VHA. As defined in VHA Directive 1200.05(3), Paragraph 3.cc, “research means a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge....” In addition, clinical investigations, including clinical investigations as defined under FDA regulations in 21 CFR 50.3, 312.3(b), and 812.3(h), are considered research as also described in the Directive. Activities that meet the definition of research under ORD policies constitute research for purposes of applicability of Section 181 of the Cleland-Dole Act. Any research activity conducted within VHA requires approval by the VA Facility’s Research and Development (R&D) Committee. This approval is in addition to any other applicable committees or subcommittees, such as an Institutional Review Board (IRB). Any VHA research activity approved by the VA Facility’s R&D Committee is excluded from the PRA. The most common types of VHA research activities to which the PRA would have previously applied (unless the research exemption was met) were research studies using collection instruments obtaining information directly from ten or more living human subjects who were not federal employees, including, but not limited to, surveys, questionnaires, or interviews. | ||
ePROS FAQ - Cleland-Dole Act Section 181Date Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Does Section 181 of the Cleland-Dole Act only apply to research conducted or funded by ORD? Answer: No, Section 181 of the Cleland-Dole Act does not apply solely to research conducted or funded by ORD. Section 181, exclusion from the PRA, applies to any research activity conducted within VHA that meets the definition of research according to ORD policies, which requires approval by the VA Facility’s R&D Committee, in addition to any other applicable committees or subcommittees. | ||
ePROS FAQ - Quality Assurance and Improvement Activities under PRADate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are quality assurance and quality improvement activities considered “research” activities that are not required to apply to PRA? Answer: No, quality assurance and quality improvement activities are not research and are required to apply the PRA if they include collections of information requiring OMB clearance under the PRA. See VHA Office of Research & Development Program Guide 1200.21 ("VHA Operations Activities That May Constitute Research") at https://www.research.va.gov/resources/policies/ProgramGuide1200-21-VHA-Operations-Activities.pdf for information about quality assurance and quality improvement activities. | ||
ePROS FAQ - Quality Improvement Research Activities under PRADate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: If a quality improvement activity uses the word “research” to describe its activities for collections of activities, is the quality improvement activity required to apply the PRA? Answer: An activity that does not meet the definition of research according to ORD policies cannot classify itself as “research” for purposes of not applying the PRA if it involves a collection of information requiring OMB clearance. For example, a quality improvement activity that does not meet the definition of a systematic investigation designed to develop or contribute to generalizable knowledge cannot be categorized as “research” when it does not meet the definition. Any research activity as defined by ORD policies require review and approval by the VA Facility’s R&D committee, including any applicable committees and subcommittees. The VA Facility’s R&D Committee cannot approve an activity as research when it is not a research activity under ORD policies. | ||
ePROS FAQ - VHA Affiliated Individuals and Entities under Section 181 of the Cleland-Dole ActDate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How are “individuals or entities affiliated with the Veterans Health Administration” defined for purposes of deciding whether the PRA applies according to Section 181 of the Cleland-Dole Act? Answer: The Cleland-Dole Act states that the PRA “. . . shall not apply to the voluntary collection of information during the conduct of research by the Veterans Health Administration, including the Office of Research and Development, or individuals or entities affiliated with the Veterans Health Administration.” The phrasing, “individuals or entities affiliated with the Veterans Health Administration” is operationalized by VHA as those individuals or entities that VHA have entered into a written agreement (e.g., contract) that are conducting the research on behalf of VHA and acting as agents of the VHA. | ||
ePROS FAQ - Federal Agency-Department defined under the Cleland-Dole ActDate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can another federal agency or department be considered “individuals or entities affiliated with the Veterans Health Administration” defined for purposes of deciding whether the PRA applies according to Section 181 of the Cleland-Dole act? Answer: If VHA is entering into a written agreement with another federal agency or department that is subject to the PRA, the specific activity in that agreement remains subject to the PRA for the non-VA federal agency or department unless the non-VA federal agency or department is conducting the research on behalf of VHA. In addition, some federal agencies, such as the National Institutes of Health (NIH), also have a PRA exemption for research activities conducted by their agencies (21st Century CURES Act, H.R. 34-114th Congress (2015-2016). https://www.congress.gov/bill/114th-congress/house-bill/34). | ||
ePROS FAQ - Updated OMB Clearance Actions under Paperwork Reduction ActDate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What actions are required by a VA Facility or VA Investigator if a VHA research activity currently has OMB clearance since the PRA no longer applies? Answer: No actions are required by a VA Facility or VA Investigator for an activity currently under OMB clearance because the PRA no longer applies under Section 181 of the Cleland-Dole Act. No documentation will be received from OMB, ORD, or the VHA PRA office. | ||
ePROS FAQ - OMB Clearance Process under PRADate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What actions are required by a VA Investigator if a VHA research activity is in the process of obtaining OMB clearance? Answer: Any activity taken by a VA Investigator to obtain OMB clearance will no longer be required to be completed because the PRA no longer applies as a result of the passage of the Cleland-Dole Act. No documentation will be received from OMB, ORD, or the VHA PRA office. If any communications are received from OMB, please send them by email to ORD (vhacoordregulatory@va.gov) and the VHA PRA office. | ||
ePROS FAQ - VHA Documentation for excluded VHA Research Activity under the Cleland-Dole ActDate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Does the Cleland-Dole Act require VHA to document when a VHA research activity is excluded from the PRA? Answer: ORD, the VHA PRA office, and/or the VA Facility’s R&D Committee are not required to document when a VHA research activity is not required to apply the PRA. Any VHA research activity that meets the definition of research as defined by ORD’s policies does not apply the PRA. | ||
ePROS FAQ - Non-Research Activity OMB Clearance under the PRADate Published: 2023-08-02 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: If the activities conducted by VHA are not a research activity, how is it determined whether the PRA applies and requires OMB clearance? Answer: Different program offices may have their own mechanism for determining whether the activity requires application of the PRA and OMB clearance. If your program does not have a mechanism, please send queries to: | ||
ePROS FAQ - ORD Clinical TrialsDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Am I required to register my ORD-funded clinical trial with ClinicalTrials.gov? Answer: Yes, access to clinical trials is critical for VA's ability to advance knowledge and care for Veterans and the nation. The VHA Office of Research and Development (ORD) is committed to informing Veterans and the public about its research and maximizing the impact of the studies it supports. To achieve these goals, PIs of ORD-funded clinical trials are responsible for registering their trial protocol and submitting summary results to the National Library of Medicine's (NLM) public registry, ClinicalTrials.gov, as a condition of funding. ORD has standardized the clinical trial registration process for trials it funds, and all trials are registered with ClinicalTrials.gov through the VA ORD Assessment and Research Reporting Tool (ART) website. Investigators use a web-based data entry form to submit all required trial information to the ART Program. The ART team then uploads the information to ClinicalTrials.gov. PIs should NOT register ORD trials outside of the ART system. ORD uses the same definition of a clinical trial as the World Health Organization. This definition is "any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes." ORD PIs should note that by applying this definition and registering a clinical trial prior to enrollment of the first participant, the trial will be eligible for publication in International Committee of Medical Journal Editors (ICMJE) member journals and other journals that follow the ICMJE requirements. For the subset of VA clinical trials that are “Applicable Clinical Trials” as defined in Section 801 of the Food and Drug Administration Amendments Act (FDAAA 801) and the Final Rule for Clinical Trials Registration and Results Information Submission (42 CFR Part 11), registering the trial through the ORD process will meet registration requirements specified in Federal law. If you are the PI of a clinical trial approved for funding and have not received an email from the ART Program with registration instructions, please contact the appropriate ORD Service representative. Note that for CSP trials, trial registration, updates and submission of study results are managed by the respective Coordinating Center. Source Document(s): | ||
ePROS FAQ - Clinicaltrials.gov - Protocol Information Update FreqencyDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How often do I need to update the protocol information on ClinicalTrials.gov? Answer: Updates to the protocol information are important for Veterans and others who are accessing ORD trial information on ClinicalTrials.gov. ORD requires PIs to update or verify the protocol information at least every 12 months while the trial is ongoing. Updates are also required by ClinicalTrials.gov. To assist PIs in this process, the ORD ART Program sends the PI email reminders when an update is due. Updates are submitted through the ART website and uploaded to ClinicalTrials.gov. In addition, for Applicable Clinical Trials, updates to specific data elements are required more rapidly under law. For example, changes to the recruitment status or the primary completion date need to be updated on ClinicalTrials.gov within 30 days of a change. The ART Program monitors specific trial data elements for all ORD trials and contacts the PI when an update may be required. Note that the protocol information can be updated at any time using the the ART website except during the summary results submission process during which changes must be made using the ClinicalTrials.gov Protocol Registration and Results System (PRS). Source Document(s): | ||
ePROS FAQ - Clinicaltrials.gov - Non-Applicable Clinical Trial Results Submission RequirementsDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: My trial does not meet the definition of an Applicable Clinical Trial, am I still required to report summary results to ClinicalTrials.gov? Answer: Yes, to help fulfill ORD’s commitment to informing Veterans and the public about its research and to maximize the impact of the studies it supports, ORD requires PIs of all clinical trials to post summary results to ClinicalTrials.gov. The requirement applies to all ORD clinical trials and is not limited to Applicable Clinical Trials as described in FDAAA 801 and the Final Rule for Clinical Trials Registration and Results Information Submission. Failure to properly report results to ClinicalTrials.gov may result in current or future project funding to be held or withdrawn by ORD. In addition, ORD will not accept or review an application from a PI who is overdue to post summary results for an ORD clinical trial. For those trials that meet the definition of an Applicable Clinical Trial, summary results must be submitted to ClinicalTrials.gov under federal regulations. Failure to submit required results information for an Applicable Clinical Trial is a prohibited act with potential legal consequences (i.e., civil or criminal judicial actions, civil monetary penalty actions) for not complying. Results are submitted using the National Library of Medicine’s ClinicalTrials.gov Protocol Registration and Results System (PRS) and not through the ORD ART website. Only protocol information is submitted and updated through ART. The ORD ART Program sends the PI an automated email notification approximately 3 months prior to the results submission due date. The email will include PRS login information and instructions for submitting summary results. To ensure timely completion, PIs should begin the process as soon as possible after receiving the email as the data entry and internal review process can take several weeks. Source Document(s): | ||
ePROS FAQ - Clinicaltrials.gov - Results Submission DeadlineDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is the required deadline for submitting trial results to ClinicalTrials.gov? Answer: Results for all ORD trials must be submitted no later than 12 months from the primary completion date. The primary completion date is defined as the date the final subject was examined or received an intervention for the purposes of final collection of data for the primary outcome measure, whether the clinical trial concluded according to the pre-specified protocol or was terminated. For those trials that meet the definition of an Applicable Clinical Trial, summary results must be submitted to ClinicalTrials.gov no later than 12 months from the primary completion date under federal regulations. Failure to submit required results information is a prohibited act under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. 331(jj)(2). Furthermore, 42 CFR 11.66 describes the potential legal consequences for responsible parties if they do not comply with the requirements to submit results information. Such potential legal consequences include civil or criminal judicial actions, civil monetary penalty actions, and grant funding actions. For all ORD trials, failure to properly report results to ClinicalTrials.gov may result in current or future project funding to be held or withdrawn by ORD. In addition, ORD will not accept or review an application from a PI who is overdue to report summary results for an ORD clinical trial. If the trial is on-going after summary results have been submitted to ClinicalTrials.gov for the primary outcome, the PI must report results for all pre-specified outcome measures or endpoints within 12 months of the study completion date. Source Document(s): | ||
ePROS FAQ - Clinicaltrials.gov - Submission ExtensionDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: May I request an extension for delayed submission of results information? Answer: Yes, PIs may request an extension of the deadline to submit clinical trial results information to ClinicalTrials.gov. PIs should direct extension requests to the appropriate ORD Service representative. For Applicable Clinical Trials, a “good cause” extension request must be submitted through the ClinicalTrials.gov Protocol Registration and Results System (PRS). The PI should first discuss the extension request with the appropriate ORD Service representative. Then, the request must be submitted to PRS prior to the date (i.e., the day before) that results information would otherwise be due. ClinicalTrials.gov does not permit responsible parties to submit good cause extension requests late (i.e., on or after the results information submission deadline). Furthermore, failure to submit required results information by the deadline is a prohibited act under the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. 331(jj)(2), for which FDA could pursue civil monetary penalties under 21 U.S.C. 333(f)(3) against the responsible party. The “good cause” extension request submitted in PRS must include a description of the reasons that constitute a good cause to justify an extension and the estimated date on which the results information will be submitted. In the request, PIs should include sufficient detail to allow for evaluation of the request. Source Document(s): | ||
ePROS FAQ - Clinicaltrials.gov - Results Submission Requirements for Publisher Accepted and Embargoed ArticlesDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: My article discussing the clinical trial results has been accepted and embargoed by the journal publisher, am I still required to submit results to ClinicalTrials.gov by the deadline? Answer: Yes, ORD requires the PI to submit summary results to ClinicalTrials.gov even when the related article has been accepted for publication. The International Committee of Medical Journal Editors (ICJME) supports clinical trial registration and transparency, and it expects authors to ensure that they have met the requirements of their funding and regulatory agencies regarding aggregate clinical trial results reporting in clinical trial registries. Editors do not consider results posted in the structured abstract and tables of the ClinicalTrials.gov registry to be “prior publication.” Source Document: ICMJE | Recommendations | Clinical Trials | ||
ePROS FAQ - Clinicaltrials.gov - Post Informed Consent DocumentDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How do I comply with the requirement to post an informed consent document to ClinicalTrials.gov? Answer: To comply with VHA Directive 1200.05 and the revised Common Rule (45 CFR 46.116(h)), PIs of ORD-funded clinical trials are required to post an informed consent document that was used to enroll participants to ClinicalTrials.gov after the trial is closed to recruitment and no later than 60 days after the last study visit by any subject (as specified in the protocol). To post an informed consent form that was used to enroll participants to ClinicalTrials.gov, redact any proprietary or personal information (e.g., names, phone numbers) from one IRB-approved informed consent form that was used to enroll patients and email the document to ART@va.gov as soon as possible after the trial is closed to recruitment. The ART Program will add the form to the clinical trial record in ClinicalTrials.gov Protocol Registration and Results System and it will be posted to the public record. Source Document(s):
| ||
ePROS FAQ - Tecovirimat EAP for MpoxDate Published: 2023-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is Required If a VA Facility Wishes to Voluntarily Close the Centers for Disease Control and Prevention’s (CDC) Tecovirimat Expanded Access Program (EAP) for Mpox at the VA Facility Under the CDC’s IRB Approval? Answer: If a VA Facility wishes to voluntarily close its participation in the CDC Tecovirimat with the CDC IRB, please email the CDC Regulatory Affairs Office at regaffairs@cdc.gov and include the answers to these questions:
Please copy the following on the VA Facility’s email to the CDC Regulatory Affairs Office:
Close the program with your VA Facility’s Research and Development (R&D) Committee after the CDC Regulatory Affairs office acknowledges the VA Facility’s email notifying them of closure. | ||
ePROS FAQ - DocuSign - Electronic Signature ComplianceDate Published: 2021-03-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is DocuSign’s electronic signature compliant with 21 CFR 11? Answer: Not all Docusign envelopes are compliant with 21 CFR 11 (aka Part 11 compliant). Only Docusign envelopes that include the Part 11 compliance module are compliant with the FDA regulations at 21 CFR 11. ORD has purchased both types of envelopes from Docusign under the IAM contract (i.e. 21 CFR 11 compliant envelopes and envelopes that are not compliant with 21 CFR 11). When submitting requests for Docusign envelopes to ORD, you will be asked to indicate whether your study is FDA-regulated (i.e., involves drugs, supplements or medical devices). If approved, studies that are FDA-regulated will be issued Docusign envelopes that are compliant with 21 CFR 11. | ||
ePROS FAQ - VA Subjects Signing Written HIPPA Authorization after Regaining CapacityDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: If an IRB or Privacy Board has approved a waiver of HIPAA authorization for subjects that do not have personal representatives (PRs), must subjects sign a written HIPAA authorization once they regain capacity? Answer: No. While there is no privacy prohibition to subjects signing a HIPAA authorization when they regain capacity, it is not required by the HIPAA Privacy Rule unless the IRB/Privacy Board-approved waiver of HIPAA Authorization specifies such limitation. It is a best practice to have the subject sign once he/she regains capacity. | ||
ePROS FAQ - Disclosing Data Containing PII and PHI Outside VA with IRB and Privacy Board Approved Waiver of HIPPA AuthorizationDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can data containing PHI/PII be disclosed outside the VA for research purposes under an IRB/Privacy Board approved waiver of HIPAA authorization? Answer: No. A waiver of HIPAA Authorization only provides legal authority to disclose PII/PHI under the HIPAA Privacy Rule. You still need authority to make the disclosure under the Privacy Act and if applicable, 38 U.S.C. 5701 and 7332. Legal authority under these other federal privacy laws may exist to permit the disclosure without the subject's signed, written authorization but that determination must be made on a case by case basis in consultation with your local Privacy Officer. | ||
ePROS FAQ - ISSO Training Requirements - CITI Training in Ethical Principles Governing Human Subjects ResearchDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are Information System Security Officers (ISSOs) required to complete Collaborative Institutional Training Initiative (CITI) training in ethical principles governing human subjects research? Answer: No. There is no requirement from ORD, Enterprise Security Operations (ESO), or Office of Information Security (OIS) for ISSOs to complete Collaborative Institutional Training Initiative (CITI) training. Facility ISSOs are required to review the ESO Research Information Security Compliance Standard Operating Procedures (SOP) and are recommended to take the following OIS-Research Support Division ITWD developed courses:
| ||
ePROS FAQ - Streamlined Process for Reviewing High-Priority-Time-Sensitive StudiesDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Does the VA Central IRB have a streamlined process for reviewing high-priority/time-sensitive research studies submitted for review? Answer: Yes. For studies requiring urgent review by the convened IRB, ad hoc meetings can be held to ensure as timely a review as possible. In addition, urgent submissions can be reviewed at a regularly scheduled meeting if there is time for the review of materials prior to the meeting. The decision to have an ad hoc meeting or to add an urgent protocol to an agenda the same week is only made by VA CIRB Administrators with the approval of the CIRB CO-Chairs for that panel. Otherwise, the submission and review process are the same for all studies. If you have a JIT deadline, please submit in a timely manner as a last minute submission will not likely be considered for urgent review. All other VA CIRB business is conducted at regularly scheduled meetings. | ||
ePROS FAQ - RCO Audit Requirement for Exempt Human Subjects ResearchDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is RCO audit required for human subjects research that is determined to be exempt from the Common Rule? Answer: Yes. ORO requires the RCO informed consent audits of exempt studies (including limited IRB review) be conducted annually. The audit requirement for exempt human research is fulfilled by completing or re-confirming the administrative data section of the ORO informed consent audit tool (or locally-modified equivalent). The box for “exempt” should be checked, and the auditor should confirm that the R&D Committee has performed initial and/or continuing reviews as required by VA policy for any active study that is not followed by any other research oversight committee (see section 2.b. of the RCO Audit Guidance). Only exempt studies that are subject to the 2018 Common Rule are required to undergo RCO regulatory audits, i.e. studies determined to be exempt on or after January 21, 2019 or exempt human subjects research activities approved by an R&DC prior to January 21, 2019 for which a VA facility has documented that the research has transitioned to the 2018 Requirements (see section 3.b. of the RCO Audit Guidance). Exempt studies that are subject to the 2018 Common Rule require a one-time completion of the administrative portion of the HRPP regulatory audit tool, as well as the “STUDY STAFF QUALIFICATIONS AND TRAINING” portion of the tool. If a study was approved by expedited review under the pre-2018 Common Rule and then transitioned to the 2018 rule and determined to be exempt, the following apply to fulfill the regulatory audit requirement of this now-exempt study: If the study had a full HRPP regulatory audit prior to its transition to the 2018 Common Rule, the study only needs to have a one-time limited/exempt HRPP audit after transition. If the study did not have a full HRPP regulatory audit prior to transition, the study needs to have a one-time full HRPP regulatory audit (look-back to study initiation) after transition. | ||
ePROS FAQ - Use of Combined ICF-HIPAA Authorization for Studies Involving BankingDate Published: 2020-06-18 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can a combined ICF/HIPAA be used if a study involves banking (mandatory or optional)? Answer: Yes. A combined ICF/HIPAA authorization can be used if a study involves mandatory banking. However, the HIPAA authorization for research cannot be combined with the informed consent document when:
REF: VHA Directive 1605.01 | ||
ePROS FAQ - Changes to Consent Form or Reconsent after Eliminating Apparent Immediate HazardsDate Published: 2020-04-06 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Do I have to change my consent form or reconsent my subjects after I implement changes to eliminate apparent immediate hazards to subjects? Answer: After prompt notification of the IRB regarding the implemented changes, either the Investigator indicates they will amend the study protocol to incorporate those changes, or the IRB will determine whether the protocol needs to be amended in order to eliminate the identified apparent hazards. The IRB also determines whether changes in the IRB-approved informed consent form are required and whether reconsenting or other notification of subjects is required. As part of the IRB approval criteria in 38 CFR 16.111(a)(4), the IRB is responsible for both the process and documentation of informed consent, including any revisions to either the process or documentation after the research is initially approved. ORD wishes to reinforce that prospective IRB approval is not required for an Investigator to implement changes to eliminate apparent immediate hazards to subjects, and that upon review of those implemented changes, the IRB needs to determine whether the protocol needs to be amended, whether the informed consent document needs to be amended, and whether previously enrolled subjects and/or currently enrolled subjects require reconsenting. The IRB may require reconsenting or an alternate means of conveying the new information to the subjects (e.g., notifying the subjects by written communication or other forms of communication as determined by the IRB and may prescribe how this communication is to be documented) | ||
ePROS FAQ - Method of Documenting Informed Consent and Eliminating Apparent Immediate HazardsDate Published: 2020-04-06 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can an Investigator make a change in the IRB-approved method of documenting informed consent prior to obtaining IRB approval for that change in order to eliminate apparent immediate hazards to human subjects? Answer: Yes. 38 CFR 16.108(a)(3)(iii) allows changes to a research activity prior to review and approval by the IRB when necessary to eliminate apparent immediate hazards to the subject. However, in all other circumstances, the IRB has regulatory authority over both the process and documentation of informed consent in a non-exempt human subjects study as described in 38 CFR§16.111(a)(4) and any changes to how informed consent documentation is obtained must be approved prospectively by the IRB. | ||
ePROS FAQ - Obtaining Written Informed Consent from Patients in IsolationDate Published: 2020-04-06 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How can written informed consent be obtained from VA research subjects who are in isolation? Answer: ORD has consulted with the Office for Human Research Protections (OHRP) and the Food and Drug Administration (FDA) about obtaining written informed consent from research subjects who are in isolation. ORD is in agreement with FDA’s guidance released on March 20, 2020 and updated on August 30, 2021 ORD entitled: FDA Guidance on Conduct of Clinical Trials of Medical Products during COVID-19 Public Health Emergency located at: https://www.fda.gov/media/136238/download. As stipulated in that guidance, the following options are available to satisfy documentation requirements when those specific methods have been approved by the IRB for obtaining written informed consent from VA patients in isolation (While the guidance was specifically addressing the COVID pandemic, the advice stands for other reasons that patients may need to be in isolation when an Investigator would like to obtain their informed consent).
If the signed informed consent document will not be able to be collected from the patient’s location and included in the study record because of contagion concerns, the following two options are acceptable to provide documentation that the patient signed the informed consent document: Option #1: Attestations by the witness who participated in the call and by the investigator that the patient confirmed that they agreed to participate in the study and signed the informed consent, Note: ORD recommends that the documented verbal confirmation include information on the version of the IRB-approved informed consent document that was used, such as IRB-approved informed consent dated 03/30/2020, Version 1.0, or other type of designation such as IRB-approved on 03/30/2020. Option #2: A photograph (or Snip & Sketch) of each page of the informed consent document with attestation by the person entering the photograph into the study record that states how that photograph was obtained and that it is a photograph of the informed consent signed by the subject. A copy of the other paper informed consent document that was signed by the investigator and witness should also be placed in the patient’s trial source documents, with a notation by the investigator of how the consent was obtained, e.g. telephone. The trial record at the investigational site should document how it was confirmed that the patient signed the consent form (i.e., either using attestation by the witness and investigator or the photographs of the signed consent). The note should include a statement of why the IC document signed by the patient was not retained, e.g., due to contamination of the document by infectious material. If the patient is unable to provide informed consent and there is a legally authorized representative, investigators should obtain consent from the participant’s legally authorized representative (only if the IRB approved the use of the subject's legally authorized representative for this study) | ||
ePROS FAQ - Modifying IRB Approved Method of Documenting Informed Consent to Eliminate Apparent HazardsDate Published: 2020-04-06 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can an Investigator make a change in the IRB-approved method of documenting informed consent prior to obtaining IRB approval in order to eliminate immediate apparent hazards to human subjects? Answer: Yes. 38 CFR 16.108(a)(3)(iii) allows changes to a research activity prior to review and approval by the IRB when necessary to eliminate apparent immediate hazards to the subject. However, in the absence of apparent immediate hazards to the subject, the IRB has regulatory authority over both the process and documentation of informed consent in a non-exempt human subjects study as described in 38 CFR§16.111(a)(4) and any changes to how informed consent documentation is obtained must be approved prospectively by the IRB.
| ||
ePROS FAQ - Biosafety - Aerosol Generating ProcedureDate Published: 2020-04-04 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is an aerosol generating procedure (AGP)? Answer: Both clinical and laboratory procedures can result in aerosol generation. A procedure that is likely to induce coughing (e.g., sputum induction, open suctioning of airways) is such a clinical procedure. These should be performed cautiously and avoided if possible. See FAQ #14 in the source document for a list of common laboratory procedures that can generate aerosols. | ||
ePROS FAQ - Biosafety - Airborne Infection Isolation RoomDate Published: 2020-04-04 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What constitutes an Airborne Infection Isolation Room (AIIR)? Answer: AIIRs are single-patient rooms at negative pressure relative to the surrounding areas, and with a minimum of 6 air changes per hour (12 air changes per hour are recommended for new construction or renovation). Air from these rooms should be exhausted directly to the outside or be filtered through a high-efficiency particulate air (HEPA) filter directly before recirculation. Room doors should be kept closed except when entering or leaving the room, and entry and exit should be minimized. Facilities should monitor and document the proper negative-pressure function of these rooms. | ||
ePROS FAQ - Multi-site Research - Employing Revised Informed Consent Documentation SimultaneouslyDate Published: 2020-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Must the revised informed consent form be employed at the same time by all sites involved in a multi-site research project? Answer: No, if multiple IRBs are involved, it is unlikely that they will all approve the document at the same time. Even if a single IRB (e.g., the VA Central IRB) is involved, release of approval letters for dozens of sites at the same time may not be practical because the forms may need to be individualized for the specific sites. (Original Post Date: March 12, 2012) | ||
ePROS FAQ - Timeline for Implementing Protocol or Informed Consent RevisionsDate Published: 2020-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How soon after IRB approval must a protocol or informed consent revision be carried out by the research team? Answer: Except when necessary to eliminate apparent immediate hazards to the subject, all changes in approved research require prospective IRB approval prior to implementation of those changes. Neither the federal regulations nor VHA policy stipulates how quickly an IRB must communicate its approval of changes to the investigator or how quickly the investigator must carry out the approved changes. However, there are some IRBs who are committed by their SOP or MOUs to provide documentation within a specific timeframe. With regards to the timeline for implementing changes, protecting the rights and welfare of the research subjects should be of primary concern in this regard. Where warranted to ensure subject safety, IRBs may stipulate that the approved changes must be implemented prior to the enrollment of new subjects and/or that previously enrolled subjects must be informed of the changes within a specified amount of time. In such circumstances, the IRB has an obligation to provide the investigator with timely notification sufficient to address the issues involved. | ||
ePROS FAQ - Use of Revised Informed Consent Form in Multi-Site ResearchDate Published: 2020-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Must the revised informed consent form be employed at the same time by all sites involved in a multi-site research project? Answer: No. If multiple IRBs are involved, it is unlikely that they will all approve the document at the same time. Even if a single IRB (e.g., the VA Central IRB) is involved, approvals of the changes at each local site may not be synchronous, and even when it is, the release of approval letters for dozens of sites at the same time may not be practical because the forms may need to be individualized for the specific sites. | ||
ePROS FAQ - Expiration Dates on Informed Consent Forms for VA ResearchDate Published: 2020-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Must IRB approval expiration dates appear on informed consent forms for VA research? Answer: No. Neither the federal regulations nor VHA policy requires that expiration dates appear on informed consent forms. One of the concerns with placing the expiration of IRB approval on the Informed consent document is the possibility a research subject might mis-intrepret that date and assume that is the date when their informed consent expires. Each IRB/HRPP has the authority to determine whether or not to require IRB approval expiration dates on the consent forms that it approves, but it is not a national requirement. | ||
ePROS FAQ - Eligibility of VA Staff for IRB Voting MembershipDate Published: 2020-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can VA ACOS/R&D, VA AO for R&D, VA research office staff, or VA IRB administrative staff serve as voting members or alternate voting members of the facility’s IRB? Answer: No. According to VHA Directive 1200.05(3), VA facility research office staff including, but not limited to, the ACOS/R&D, the AO for R&D, and IRB administrative staff may not serve as voting members, or alternate voting members, of the facility’s IRB of record. | ||
ePROS FAQ - Institution Specific Form TemplatesDate Published: 2020-03-03 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Will each institution have their own specific templates for their forms? Answer: ORPP&E, along with a field-based workgroup, has developed a core library of form and letter templates for each of the committee and subcommittee workspaces in IRBNet. However, IRBNet supports the ability for you to upload your own supplemental forms (such as if a particular form or template is required to support state or local regulations) via the Library Manager. | ||
ePROS FAQ - IRBNet Integration with ePROMISEDate Published: 2020-03-03 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Will IRBNet update the station's ePROMISE records? Answer: Currently the system does not integrate with ePROMISE. We hope to integrate VAIRSS with other ORD enterprise systems in the near future. | ||
ePROS FAQ - Azure RMS - Large Data File TransferDate Published: 2019-10-25 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can Azure RMS be used by VA researchers to send large data files to another researcher? Answer: Azure RMS can transfer files up to 40Mb. | ||
ePROS FAQ - Utilizing Azure RMS for VA Subjects Communication with PII and PHIDate Published: 2019-10-25 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can Azure RMS be used by VA researchers to communicate personally identifiable information and protected health information (PII/PHI) to VA subjects? Answer: Yes. Azure RMS can be used to communicate PII/PHI to VA subjects as part of the approved VA research protocol. External Recipients can open or view RMS encrypted email from any compatible web browser or through their social email account (Gmail, Yahoo, Outlook). | ||
ePROS FAQ - Utilizing Azure RMS for Research Collaborator CommunicationDate Published: 2019-10-25 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can Azure RMS be used by VA researchers to send a secure email to a research collaborator? Answer: Yes. Azure RMS can be used to send a secure email to a research collaborator as part of the approved VA research protocol. Azure RMS provides encryption through email for compliance with Federal Information Protection Standards (FIPS) 140-2 and other VA Sensitive Information (VASI) compliance requirements. Official VA Research communication must be initiated through a VA Outlook account using VA OI&T approved Azure RMS Procedures. | ||
ePROS FAQ - Considerations for IRB or VA RD Committee Evaluation of Studies Utilizing Azure RMS for CommunicationDate Published: 2019-10-25 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What are some considerations for an IRB or VA R&D Committee evaluating a research study proposing to utilize Azure RMS to recruit or communicate messages containing PII/PHI with VA subjects? Answer: Both exempt and non-exempt human subjects studies may be conducted using Azure RMS to recruit or communicate messages containing PII/PHI with VA subjects. The use of Azure RMS is analogous to any method of communication used in human subjects research; its use must be evaluated by the applicable research oversight committee(s). If standardized communications are used, the applicable research oversight committee must review the content of standardized communications. Other considerations for the use of Azure RMS to recruit or communicate messages containing PII/PHI include, but are not limited to, the following: | ||
ePROS FAQ - Including Azure RMS in Informed Consent DocumentationDate Published: 2019-10-25 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: For VA research requiring a written informed consent document, is the IRB required to include specific language about use of Azure RMS? Answer: ORD does not have a policy requiring that Azure RMS must be described in the written informed consent approved by an IRB. However, the IRB has authority to determine the content of informed consent to ensure that it is in accordance with, and to the extent required by 38 CFR 16.116. The Common Rule (e.g., the Federal Policy for the Protection of Human Subjects codified by VA as 38 CFR Part 16) requires as part of the basic elements of informed consent in 38 CFR Part 16.116(b)(1) that the following be provided to each subject or the subject’s legally authorized representative: | ||
ePROS FAQ - Replacement of Required Approvals or Waivers for Non-Veteran Inclusion after RD Committee ApprovalDate Published: 2019-09-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: If the R&D Committee approves the inclusion of non-Veterans in an ORD-funded study, does it replace any required approvals or waivers for inclusion of non-Veterans by the ORD funding service? Answer: No, R&D committee approval of inclusion of non-Veterans in an ORD-funded study does not replace any requirements of the applicable funding service. | ||
ePROS FAQ - Reportable Events for Exempt Human Subjects ResearchDate Published: 2019-08-07 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Which committee is responsible for reportable events involving Exempt human subjects research? Answer: VHA Directive 1058.01 only requires prompt reporting on non-exempt human subjects research. Given several categories of exempt human subject research require Limited IRB Review (and approval), prompt reporting of noncompliance or UPIRTSOs relating to the safequarding of subjects privacy and confidentiality rights should go back to the IRB that conducted the Limited IRB review. Any other required reporting would only follow from local SOPs and would go to the RDC as this is the primary oversight body for exempt human subjects research. | ||
ePROS FAQ - Non-VeteransDate Published: 2019-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Why is the R&D Committee required to review and approve recruitment of non-Veterans in VA research instead of an Institutional Review Board (IRB)? Answer: VA research focuses on health issues that affect Veterans. A Veteran as defined in 38 U.S. Code §101(2) “. . . means a person who served in the active military, naval, or air service, and who was discharged or released therefrom under conditions other than dishonorable.” The R&D Committee is responsible for ensuring that all research in which the facility is engaged is consistent with the VA mission. The evaluation of whether the inclusion of non-Veterans in a proposed VA research activity is consistent with meeting the VA mission cannot be delegated to an Institutional Review Board (IRB) because the evaluation is not a human subjects protections issue; it is an institutional evaluation. In addition, VA conducts many exempt human subjects research activities that could involve non-Veterans. Not all exempt human subjects research activities require IRB approval. Common examples of exempt human subjects research activities involving non- Veterans are survey research involving caregivers and employees. | ||
ePROS FAQ - Status of VA Central IRB as a Subcommittee of the RD CommitteeDate Published: 2019-06-05 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is the VA Central IRB a subcommittee of the R&D Committee? Answer: No. The VA Central IRB is not a subcommittee of the R&D Committee. While it is a VA IRB, and therefore an internal IRB, with regards to committee status. it is an external committee established by a Memorandum of Understanding (MOU) between a VA Facility and Central Office. As stated in the Note in VHA Directive 1200.01(1), Paragraph 8.a., “External committees established by MOUs or other agreements in lieu of required subcommittee(s) are not considered subcommittees and are governed by the agreement (e.g. the VA Central IRB).” | ||
ePROS FAQ - Classification of VA Central IRB as an Internal or External VA IRBDate Published: 2019-05-24 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is the VA Central IRB an internal IRB or an external IRB? Answer: The VA Central IRB is an internal IRB. As stated in the Note in VHA Directive 1200.01(1), Paragraph 5.h.(7): “For purposes of this directive, use of the VACO IRB or another VA facility’s internal IRB is not considered to be an external IRB." Internal IRBs include two types of IRBs:
| ||
ePROS FAQ - RD Committee Continuing Review Requirement of Activities Approved by Expedited Review or the Common RuleDate Published: 2019-05-24 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Is the R&D Committee required to conduct continuing review for human subjects research activities approved by expedited review or transitioned to the 2018 Requirements of the Federal Policy for the Protection of Human Subjects (Common Rule) when the IRB does not conduct continuing review of the research activity? Answer: No, the R&D Committee is not required to conduct continuing review of the non-exempt human subjects research activities because the research activities remain under the continuing oversight of the IRB. | ||
ePROS FAQ - My HealtheVet InformationDate Published: 2018-03-12 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is My HealtheVet? Answer: My HealtheVet is an online portal created by the Department of Veterans Affairs to help Veterans, active-duty service members, and their dependents work with healthcare providers to reach informed decisions and improve their overall health. My HealtheVet offers a suite of tools designed to assist Veterans in managing their health care. VA patients with a Premium My HealtheVet account can access their health records, refill VA prescriptions, receive email reminders about appointments, and communicate with members of their healthcare team and other VA staff using Secure Messaging. | ||
ePROS FAQ - VA Subjects Communications - Email and Text Message RetentionDate Published: 2017-07-28 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are VA Investigators required to keep copies of emails and text messages sent to and from VA subjects? Answer: Emails and text messages sent and received by a VA Investigator and the VA research team are federal records subject to the VHA Record Control Schedule. Copies of the email communications and text messages sent and received by a VA Investigator and the VA research team must be maintained in accordance with the VHA Record Control Schedule (RCS 10-1) as part of local VA Investigator records. | ||
ePROS FAQ - Utilization of My HealtheVets Secure Messaging SystemDate Published: 2017-07-28 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can VA Investigators utilize My HealtheVet’s Secure Messaging system to recruit VA subjects in approved VA research studies? Answer: VA Investigators cannot use My HealtheVet’s Secure Messaging system to recruit VA subjects. | ||
ePROS FAQ - Email Recruitment and Communication with VA SubjectsDate Published: 2017-07-28 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Can email messages be sent by VA Investigators to recruit and communicate with VA subjects? Answer: Yes. When meeting the specific requirements below, VA Investigators can use VA email to recruit prospective VA subjects and to communicate with VA subjects who have consented to participate in a VA research study as described in the IRB-approved research study. VA Investigators may not utilize their personal email accounts (e.g., Google) or university email accounts for research communications with prospective or consented VA subjects. The use of personal email account or the use of a personal email system to conduct official agency business is not allowed. No PII/PHI can be sent by a VA Investigator for VA research purposes to a prospective or consented VA subject by email unless the email is encrypted using a VA-approved encryption method. Your local ISO should review the encryption to ensure it meets all applicable requirements. If the message is not encrypted, ORD recommends that the email message be reviewed as part of IRB review process. The IRB should consult with the VA Facility’s Privacy Officer concerning privacy issues outside the scope of the human subject protection regulations. Note: Azure Rights Management System (RMS)can be used by VA researchers to send a secure email to communicate personally identifiable information and protected health information (PII/PHI) to VA subjects. Additional guidance on Azure RMS is available here: https://www.research.va.gov/resources/policies/guidance/FAQs-Azure-RMS.pdf | ||
ePROS FAQ - IRB Membership and the Non-Affiliated MemberDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Who is a "non-affiliated individual"? Answer: The regulatory requirements for IRB membership includes "at least one member who is not otherwise affiliated with the institution and who is not part of the immediate family of a person who is affiliated with the institution." This means that the person has no discernable ties, ongoing relationship, or association with the VA. An immediate family member (parent, spouse, child, sibling) of an affiliated person may not be considered a “non-affiliated individual.” Note: Veterans whose only relationship with VA is receiving care at a VA facility or receiving benefits from the Veterans Benefits Administration are not considered to be “affiliated” for the purpose of being an IRB member. Employees of the academic affiliate are considered non-affiliated for purposes of determining membership eligibility on a VA IRB if they do not otherwise have an appointment (i.e., compensated, Intergovernmental Personnel Act (IPA), or WOC). Individuals who perform occasional volunteer activities at VA without a WOC appointment are not considered “affiliated” for purposes of this Guidance document. However, those who hold a WOC appointment for volunteer activities other than IRB service are considered to be affiliated. Individuals who have retired from VA are considered affiliated for purposes of determining membership eligibility on a VA IRB only if they are receiving VA retirement benefits. Individuals who receive a WOC appointment solely for the purpose of their appointment to the IRB as a not otherwise affiliated member are not considered to be otherwise affiliated. | ||
ePROS FAQ - Academic Affiliation and IRB MembershipDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What does academic “affiliation” mean for the purposes of VA IRB Membership? Answer: An “academic affiliate” means an academic institution that has a relationship for the purpose of education, research, or enhanced patient care with a VA facility documented by an executed Affiliation Agreement in conformance with VA requirements. In addition, special purpose agreements documented by a memorandum of understanding approved by the Chief Research and Development Officer may be developed in research and development (R&D) areas, such as health services or rehabilitation R&D. | ||
ePROS FAQ - Employees of Academic Affiliates and IRB MembershipDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are employees of the academic affiliate considered affiliated with VA with regards to IRB Membership? Answer: Employees of the academic affiliate are considered non-affiliated for purposes of determining membership eligibility on a VA IRB if they do not otherwise have an appointment (i.e., compensated, Intergovernmental Personnel Act (IPA), or WOC) at the VA, or any other discernable ties, ongoing relationship, or association with the VA. | ||
ePROS FAQ - IRB Membership and Veterans Receiving Care at VADate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are Veterans receiving care at the VA considered to be affiliated for purposes of IRB membership? Answer: Veterans whose only relationship with VA is receiving care at a VA facility or receiving benefits from the Veterans Benefits Administration are not considered to be “affiliated” for the purpose of being an IRB member. | ||
ePROS FAQ - IRB Membership and Veterans Who Volunteer at the VADate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are Veterans who volunteer at the VA considered to be affiliated for purposes of IRB membership? Answer: Individuals who perform occasional volunteer activities at VA without a WOC appointment are not considered “affiliated” for purposes of this Guidance document. However, those who hold a WOC appointment for volunteer activities other than IRB service are considered to be affiliated. | ||
ePROS FAQ - IRB Membership and VA RetireesDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are VA retirees considered to be affiliated for purposes of IRB membership? Answer: Individuals who have retired from VA are considered affiliated for purposes of determining membership eligibility on a VA IRB only if they are receiving VA retirement benefits. | ||
ePROS FAQ - What are WOC AppointmentsDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What does receiving a WOC appointment mean? Answer: A WOC is a "Without Compensation" designation for an individual who has an official VA appointment but does not receive any salary or benefits from VA (38 U.S.C. §§ 513, 7405(a)(1)). This appointment may allow the individual to support VA’s research program in various capacities including, but not limited to, being an investigator, research coordinator, and administrator while at VA for a defined period of time. WOCs are subject to all laws and regulations pertaining to government personnel, including, but not limited to, government ethics laws and standards of conduct and VHA’s credentialing and privileging policy. A WOC is considered a VA employee. Note: A WOC may be an employee of a university affiliate or other nonprofit organization and is not under an IPA. An IPA is also a VA appointment that allows an individual to serve as a VA employee, therefore, an IPA does not require a WOC appointment. | ||
ePROS FAQ - WOC Appointment for Nonaffiliated IRB MembersDate Published: 2015-04-16 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Should a nonaffiliated IRB member be given a WOC appointment? Answer: A nonaffiliated VA IRB member should, but is not required to, obtain a WOC appointment. The WOC appointment provides the best protection from unexpected liability and the best likelihood for the Department of Justice (DOJ) to represent that individual as a VA "employee" and thus, be immune from personal liability for negligence. The WOC appointment for purposes of liability coverage does not change the affiliation status of the IRB member. The WOC appointment may also be required for the individual to obtain a PIV card, and laptop computer in order to access VAIRRS. NOTE: If a nonaffiliated IRB member chooses not to obtain a WOC appointment for purposes of being on the IRB, he/she is still bound to all VA requirements on confidentiality and conflict of interest. See additional guidance available at www.research.va.gov/resources/policies/guidance/NonAffiliated-IRB.docx | ||
ePROS FAQ - Requirements for Collecting Pregnancy Information in VA ResearchDate Published: 2015-03-09 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is required when collecting Pregnancy Information from or about Female Subjects who become Pregnant while Participating in VA Research Answer: In the majority of studies conducted in the VA, pregnancy is not the focus of the research. In many research studies, particularly clinical investigations involving interventions with drugs or medical devices, the investigator or sponsor may want to obtain information about the progress of a pregnancy when a research subject, or subject's partner becomes pregnant during the subject's participation in the study. In the many investigations, if the subject becomes pregnant (and sometimes if the partner becomes pregnant) the protocol will require the the clinical intervention to end for the subject, but ongoing monitoring of the pregnant partner and fetus may be sought for understanding the possible risks related to pregnancy. If a subject who becomes pregnant is withdrawn from the research intervention portion of the study by a VA investigator, collection of any additional data by the investigator from the pregnant subject (or partner) after the subject has been withdrawn would constitute human subject research activities. A subject can be withdrawn solely from receiving the research intervention, remain in the study, and the investigator may continue data collection and other follow-up activities if the data collection and follow-up activities are described in the protocol and informed consent approved by the IRB, the HIPAA authorization, and the subject does not revoke that portion of the informed consent and HIPAA authorization. Written authorization requirements for use and disclosure of protected health information must be met as described in VHA Directive 1605.01 ("Privacy and Release of Information") and VHA Directive 1200.05(3). When a subject is withdrawn from all aspects of the study by either the subject's or investigator's choice, the subject's participation in that study ends and additional data and identifiable private information about that subject must not be collected or obtained. | ||
ePROS FAQ - Collecting Pregnancy Information from Female Partners of Male Subjects in VA ResearchDate Published: 2015-03-09 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is required when Collecting Pregnancy Information from or about Female Partners of Male Subjects Participating in VA Research Answer: The majority of studies conducted in VA involve recruitment of male subjects. In research studies, particularly clinical investigations, the investigator may want to obtain information about the progress of a pregnancy if the female partner of a male Veteran subject becomes pregnant during the interval when the male subject is participating in a VA research study. VA applies the Common Rule to all human subjects research which is approved as VA research. Collection of data in the research through intervention or interaction with the female partner or obtaining identifiable private information about the female partner constitutes human subjects research. The female partner becomes a human subject according to the Common Rule and is given protections as a human subject as required in 38 CFR §16 and VHA Directive 1200.05(3). In such cases, the IRB-approved protocol must include provisions for collecting information from the pregnant female partner of the male subject enrolled into the research. Informed consent as approved by the IRB must be obtained by the investigator from the female partner prior to the collection of any data by the investigator from the female partner or obtaining identifiable private information about her unless the IRB has waived informed consent in accordance with criteria described in 38 CFR § 16.116(c) or 38 CFR §16.116(d). Written authorization for use and disclosure of protected health information must be obtained from the female partner as described in VHA Directive 1605.01: Privacy and Release of Information. | ||
ePROS FAQ - Collecting Information About Newborns in VA ResearchDate Published: 2015-03-09 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What is required when collecting Information about the newborn in VA Research? Answer: When a VA investigator conducting human subjects research collects identifiable private information about the newborn infant of subject enrolled in VA research, the infant is a human subject, and the investigator is conducting VA research involving children. The IRB-approved protocol must include provisions for collecting information about the newborn infant, and the IRB needs to have met the requirements for reviewing research involving children in accordance with VHA Directive 1200.05. Informed consent/parental permission as approved by the IRB and obtained from the adult subject(s) must contain sufficient information about the data to be obtained about the newborn. Written authorization for use and disclosure of protected health information must also be obtained as described in VHA Directive 1605.01: Privacy and Release of Information. Finally, the VA facility director must have approved the participation of children in the research prior to collection of data about the newborn infant following IRB review and approval in accordance with VHA Directive 1200.05(3) requirements. | ||
ePROS FAQ - IRB Considerations for Collecting Pregnancy Data in VA ResearchDate Published: 2015-03-09 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What should the IRB consider when collecting data on pregnancy progress and outcomes of pregnancy in VA Research? Answer: When VA research involves collecting data from or about pregnancy and pregnancy outcomes, the IRB must still ensure that subjects' ethical rights are protected. VHA follows 45 CFR §46.204 requirements for including pregnant women in VA research. IRBs are not expected or required to review studies using the criteria described in 45 CFR §46.204 simply because the study involves women of child-bearing potential. However, once the reviewing IRB is made aware that information about a subject's pregnancy is being obtained in a VA study, the reviewing IRB must ensure that:
| ||
ePROS FAQ - Determining the Frequency of Continuing Review by IRBDate Published: 2014-10-22 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: How does the IRB determine the frequency for continuing review? Answer: When continuing review is required by the IRB, it must be conducted at intervals appropriate to the degree of risk but not less than once per year. The IRB should decide the period of approval (which is also the interval for continuing review) for each research project in order to ensure the continued protection of the rights and welfare of research subjects. More frequent review (i.e., more frequently than once per year) may be appropriate when the risks to subjects, or other circumstances involved in the research warrant more frequent reassessment. Other factors may include a new Investigator, or a high risk study. The IRB should consider factors such as the following when deciding on an appropriate interval for continuing review:
In addition to specifying a time interval, the IRB may specify a subject enrollment number as a threshold for determining when continuing review is to occur (e.g., at the time of initial review and approval of a high-risk clinical trial, the IRB might require that continuing review occur either in 6 months or after 2 subjects have been enrolled, whichever occurs first). | ||
ePROS FAQ - IRB Review Criteria for Continuing ReviewDate Published: 2014-10-22 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What does the IRB review during continuing reviews? Answer: Continuing review is an opportunity for the IRB to reconsider the criteria for approval as they relate to the study with additional information and experience with applyinig study interventions to the research sample. The IRB reviews all of the information submitted and determines whether the criteria for approval are still met:
The IRB should determine if there is any new information provided by the investigator, or otherwise available to the IRB, that would alter the IRB’s prior determinations, particularly with respect to the IRB’s prior evaluation of the potential benefits or risks to the subjects, and to the adequacy of the informed consent information and documentation. If the research at continuing review does not satisfy all of the above criteria, the IRB must require changes that would result in the research satisfying these criteria, defer taking action, or disapprove the research. | ||
ePROS FAQ - IRB Continuing Review Using Expedited ProceduresDate Published: 2014-10-22 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: When can the IRB use expedited procedures for continuing review? Answer: A research study that was eligible for initial review under an expedited review procedure (met one of the expedited review categories listed at: https://www.hhs.gov/ohrp/regulations-and-policy/guidance/categories-of-research-expedited-review-procedure-1998/index.html, and involved no more than minimal risk) and which was required to undergo a continuing review, will qualify for an expedited review procedure at the time of continuing review. This assumes that there have not been changes to the research that include components that fall outside of the expedited review categories, and that there is not new information available so that the IRB no longer determines the research to be no more than minimal risk. Under the 2018 Common Rule provisions, no minimal risk study is required to undergo continuing review unless the IRB requires it and documents its rational for doing so. Research projects not initially eligible for expedited review may become eligible for expedited continuing review under the following circumstances:
| ||
ePROS FAQ - Consequences of a Lapse in IRB ApprovalDate Published: 2014-10-22 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What happens if there is a lapse of IRB approval? Answer: The conduct of human subjects research is only permissible with IRB approval based on federal regulations and VHA policy (Directive 1200.05(3)). If continuing review is required, and has not been completed and/or the study has not been re-approved by the end of any IRB approval period, the study lapses in approval. A lapse in IRB approval can be considered serious noncompliance with the regulations and with the requirements of the IRB. If approval expires:
A lapse in IRB approval is not a suspension or termination of IRB approval. Therefore, such expirations of IRB approval do not need to be reported to ORO or OHRP. However, if the IRB notes a pattern of non-compliance with the requirements for continuing review (e.g., an investigator repeatedly or deliberately neglects to submit materials for continuing review in a timely fashion), the IRB should determine whether such a pattern represents serious or continuing noncompliance that needs to be reported to appropriate institutional officials (see VHA Directive 1058.01). | ||
ePROS FAQ - Conducting Research Involving Pregnant Women in VADate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Under what circumstances can research involving pregnant women be conducted in VA? Answer: Research involving pregnant women may be conducted in VA under the following circumstances:
| ||
ePROS FAQ - Required Information for Research Proposals Involving Pregnant WomenDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What information should be provided in a research proposal and/or review materials that involve pregnant women in research? Answer: The following should be provided for review of research involving pregnant women:
| ||
ePROS FAQ - IRB Review and Documentation for Approving Research Involving Pregnant WomenDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What must the VA IRB of record review and document for approving research involving pregnant women? Answer: When approving research involving pregnant women, the VA IRB of record must document:
| ||
ePROS FAQ - Research Involving Children - RD Committee Review and Documentation RequirementsDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What should the R&D Committee review and document for research involving children? Answer: The proposed research is relevant to the VA’s mission and the care of Veterans.
| ||
ePROS FAQ - Research Involving Children - Facility Director Memo of Approval RequirementsDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What are the requirements for the facility Director memo of approval for research involving children? Answer: CRADO approval is not required for a VA investigator to conduct VA research involving children. However, the facility Director should approve the conduct of such research before it is initiated.
| ||
ePROS FAQ - Conditions for Conducting Research Involving Children at the VADate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Under what circumstances can research involving children be conducted in VA? Answer: Research involving children may be conducted at VA when:
| ||
ePROS FAQ - Required Information for Research Proposals Involving ChildrenDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What information should be provided in a research proposal and/or review materials that involve children in research? Answer: The following information should be provided in materials submitted for review of research involving children.
Documentation of any additional safeguards that have been incorporated into the clinical or research site where children will be studied.
| ||
ePROS FAQ - IRB Review and Documentation for Approving Research Involving ChildrenDate Published: 2014-10-20 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: What should the VA IRB of record review and document for approving research involving children? Answer: The VA IRB of record should review and document the following when approving research involving children:
| ||
ePROS FAQ - Conflict of InterestDate Published: 2013-02-08 00:00:00.0 Recertification Date: 2024-08-15 00:00:00.0 Owner: ePROS
Question: Are Public Health Service (PHS) regulations regarding investigator financial conflicts of interest (FCOI) that went into effect August 24, 2012, applicable to VA research? Answer: No. The PHS regulations are found at 42 CFR 50.601-607 (especially 42 CFR 50.604, “Responsibilities of Institutions regarding Investigator financial conflicts of interest”) and do not apply to VHA. Although the PHS regulations state they are applicable to “each institution that is applying for, or that receives, PHS research funding …,” (42 CFR 50.601), the definition of “institution” excludes Federal agencies (42 CFR 50.603). |